The goal of pajam is to provide data and visualizations from Protein Atlas, whose data is graciously provided by https://proteinatlas.org.
Installation
You can install pajam from GitHub with:
# install.packages("remotes") remotes::install_github("jmw86069/pajam")
Package Reference
Online documentation is available via pkgdown:
Full pajam function reference: https://jmw86069.github.io/pajam
Examples
Display protein tissue expression for a set of genes, displaying only "Tissue" sample types:
library(pajam) test_genes <- c("DKK1","DKK4","CXCL12","IL6R","MET", "HK2","FTL","FTH1","STAT1","STAT3","CDKN1B"); rowGroupMeans <- jamba::rowGroupMeans; proteinatlas_heatmap(genes=test_genes, type="Tissue", centered=TRUE);

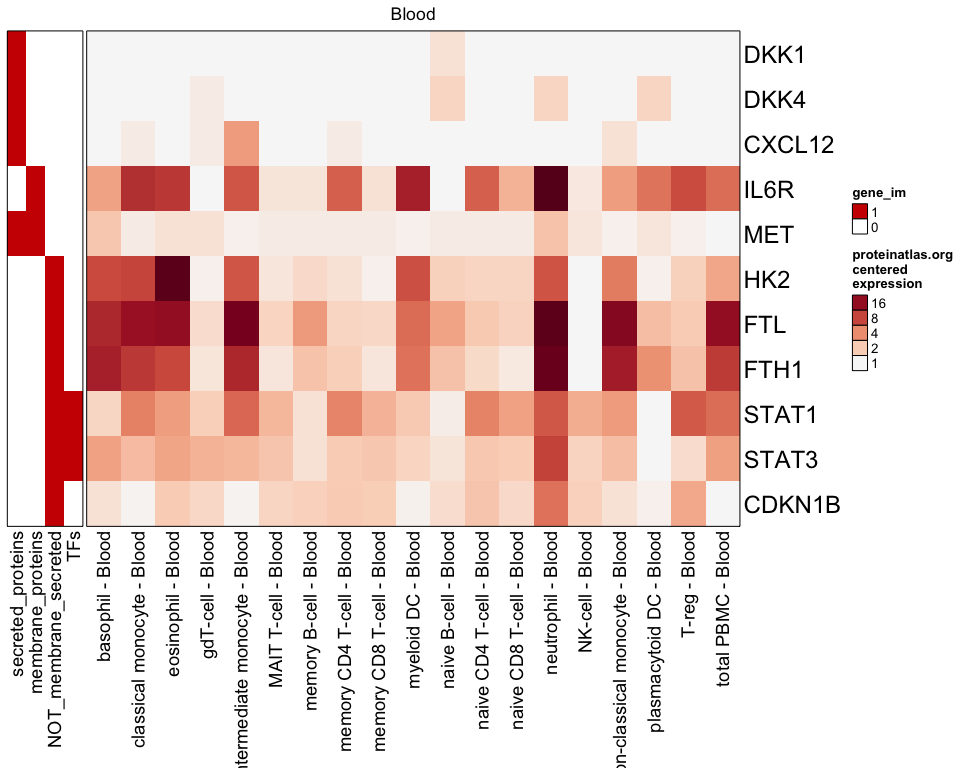
This package includes a few useful genesets that may provide useful context into the genes. The following example shows "Blood" sample types, and some added annotations.
# use proteinatlas_genesets_fdb11 use_im <- c("secreted_proteins", "membrane_proteins", "NOT_membrane_secreted", "TFs"); proteinatlas_im <- list2im_opt(proteinatlas_genesets_fdb11[use_im]); proteinatlas_heatmap(genes=test_genes, type="Blood", centered=TRUE, gene_im=proteinatlas_im);
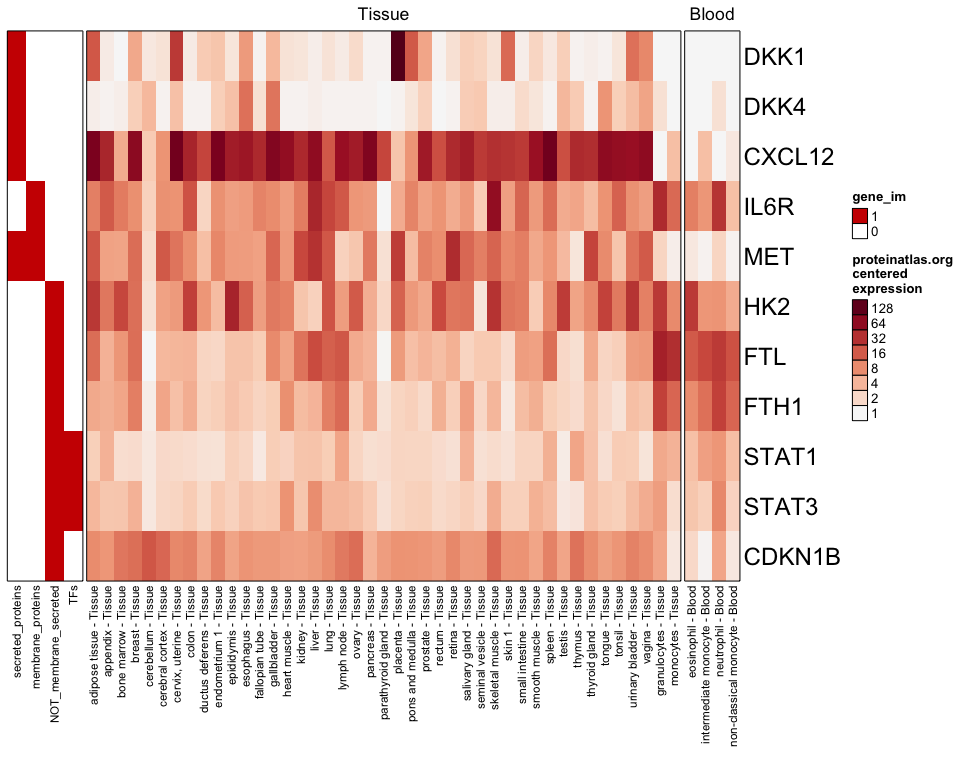
You can filter samples to require a certain minimum expression:
proteinatlas_heatmap(genes=test_genes, type=c("Tissue","Blood"), centered=TRUE, column_filter=16, gene_im=proteinatlas_im);
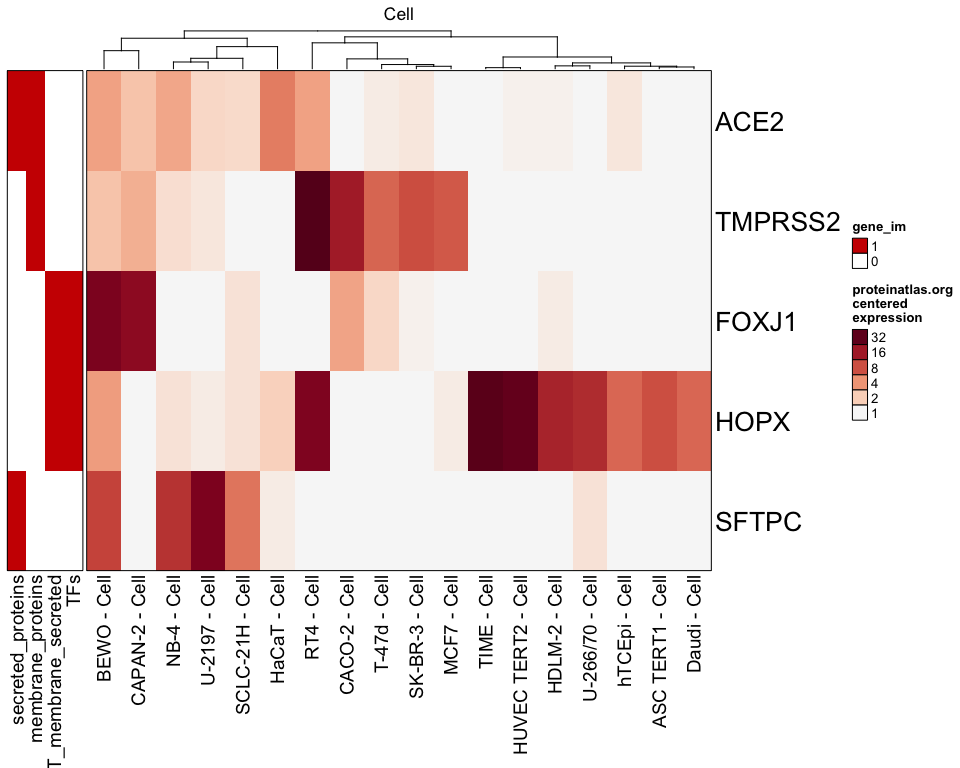
It may be useful in finding cell line expression for a subset of genes:
proteinatlas_heatmap(genes=c("ACE2", "TMPRSS2", "FOXJ1", "HOPX", "SFTPC"), type=c("Cell"), centered=TRUE, column_filter=4, cluster_columns=TRUE, gene_im=proteinatlas_im);
R-shiny app
You can start an R-shiny app that includes a heatmap that can be zoomed (thanks to the ComplexHeatmap package!):
## not run #launch_pajam(); # Or define custom starting genes selected_genes <- c("DKK1","DKK4","CXCL12","IL6R","MET", "HK2","FTL","FTH1","STAT1","STAT3","CDKN1B"); #launch_pajam();