Import genome coverage matrix files
coverage_matrix2nmat(
x = NULL,
filename = NULL,
signal_name = NULL,
target_name = "target",
background = 0,
smooth = FALSE,
target_is_single_point = FALSE,
signal_is_categorical = FALSE,
mat_grep = "[-0-9]+:[-0-9]+",
upstream_grep = "^[-]",
downstream_grep = "^[^-]",
target_grep = "^0$",
verbose = FALSE,
...
)Arguments
- x
data.frameor compatible object containing genome coverage data, or a character file path. Whenxis not supplied,filenameis used to import data. Whenxis a filename, it is used to populatefilename, then data is imported intox.- filename
character path to a genome coverage file. When
xis supplied, this argument is ignored. Whenfilenameis used, only the first file is imported.- signal_name
The name of signal regions. It is only used for printing the object. When
signal_nameisNULL, thesignal_nameis derived fromnames(filename)if available, thenbasename(filename), or"signal"then onlyxis supplied.- target_name
The name of the target names. It is only used for printing the object.
- background
numeric value containing the background value in the matrix.
- smooth
logical whether to apply smoothing on rows.
- target_is_single_point, signal_is_categorical
logical indicating whether the target region is a single point, and whether signal matrix is categorical, respectively.
- mat_grep
character regular expression pattern used to identify colnames which contain coverage data. The default pattern expects the format
"-200:-100".- upstream_grep
character regular expression pattern used to identify upstream colnames from values that match
mat_grep. The default assumes any region beginning"-"is negative and upstream the central target region.- downstream_grep
character regular expression pattern used to identify upstream colnames from values that match
mat_grep. The default assumes all colnames which are not upstream are therefore downstream.- target_grep
character regular expression pattern used to identify a colname referring to the
target, which by default can only be"0". Otherwise, no target region is defined.- verbose
logical indicating whether to print verbose output.
- ...
additional arguments are ignored.
Value
normalizedMatrix numeric matrix, where additiona
metadata is stored in the object attributes. See
EnrichedHeatmap::as.normalizedMatrix() for more
details about the metadata. The rownames are defined
by the first colname which does not match
mat_grep, which by default is "Gene ID",
otherwise rownames are NULL.
Details
This function imports genome coverage data matrix
and returns an object of class
normalizedMatrix compatible for use by the
package "EnrichedHeatmap".
There is a conversion function EnrichedHeatmap::as.normalizedMatrix(),
however this function does not call that function, in
favor of defining the attributes directly. In future, this
function may change to call that function.
See also
Other jam coverage heatmap functions:
get_nmat_ceiling(),
nmathm_row_order(),
nmatlist2heatmaps(),
validate_heatmap_params(),
zoom_nmatlist(),
zoom_nmat()
Other jam import functions:
deepTools_matrix2nmat(),
frequency_matrix2nmat(),
import_lipotype_csv(),
import_metabolomics_niehs(),
import_nanostring_csv(),
import_nanostring_rcc(),
import_nanostring_rlf(),
import_proteomics_PD(),
import_proteomics_mascot(),
import_salmon_quant(),
process_metab_compounds_file()
Examples
## There is a small example file to use for testing
cov_file <- system.file("data", "tss_coverage.matrix", package="platjam");
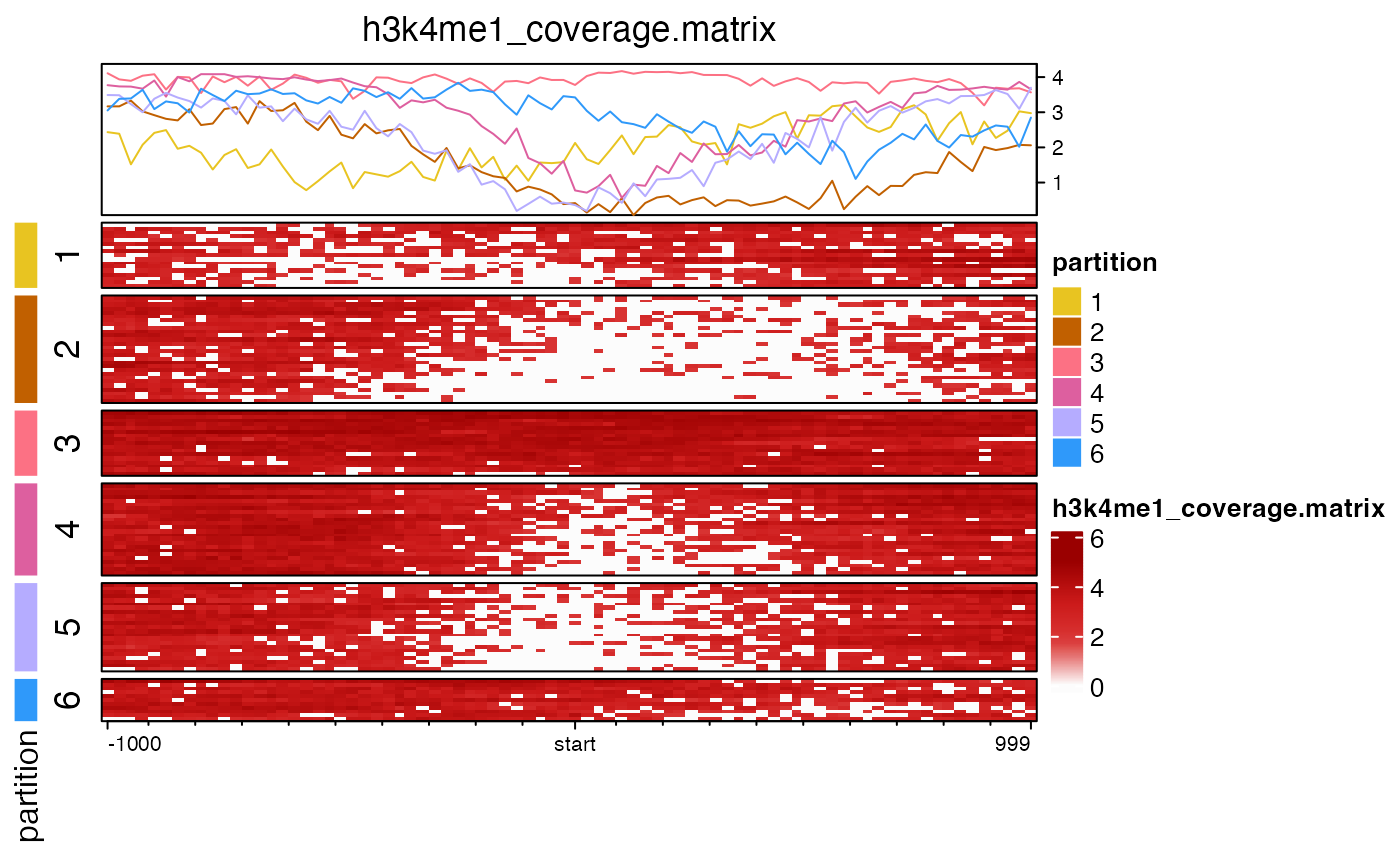
cov_file <- system.file("data", "h3k4me1_coverage.matrix", package="platjam");
if (length(cov_file) > 0) {
nmat <- coverage_matrix2nmat(cov_file);
jamba::printDebug("signal_name: ",
attr(nmat, "signal_name"));
if (suppressPackageStartupMessages(require(EnrichedHeatmap))) {
color <- "red3";
signal_name <- attr(nmat, "signal_name");
k <- 6;
set.seed(123);
partition <- kmeans(log10(1+nmat), centers=k)$cluster;
EH <- EnrichedHeatmap(log10(1+nmat),
split=partition,
pos_line=FALSE,
use_raster=TRUE,
col=jamba::getColorRamp(color, n=10),
top_annotation=HeatmapAnnotation(
lines=anno_enriched(gp=grid::gpar(col=colorjam::rainbowJam(k)))
),
axis_name_gp=grid::gpar(fontsize=8),
name=signal_name,
column_title=signal_name
);
PHM <- Heatmap(partition,
use_raster=TRUE,
col=structure(colorjam::rainbowJam(k),
names=as.character(seq_len(k))),
name="partition",
show_row_names=FALSE,
width=grid::unit(3, "mm"));
draw(PHM + EH, main_heatmap=2);
}
}
#> ## (12:31:34) 21Sep2023: signal_name: h3k4me1_coverage.matrix