Sample exon data GRanges with wide introns
Format
GRanges object where each segment represents one
exon for an arbitrary gene. It has one column of values,
"gene_name" used for Sashimi plot preparation.
Details
This dataset contains RNA-seq splice junction data stored as a GRangesList.
Intron and exon sizes are more consistent with mammalian gene structure, and are intended to demonstrate the challenge with visualizing exon coverage data on a genomic scale. See examples for steps to compress the intron sizes.
See also
Other Splicejam data:
sjenvtest,
test_cov_gr,
test_cov_wide_gr,
test_exon_gr,
test_junc_gr,
test_junc_wide_gr
Examples
# The code below is used to create the exon test data
suppressPackageStartupMessages(library(GenomicRanges));
suppressPackageStartupMessages(library(ggplot2));
test_exon_wide_gr <- GRanges(seqnames=rep("chr1", 4),
ranges=IRanges::IRanges(
start=c(100, 10300, 20500, 39900),
end=c(200, 10400, 20750, 40000)),
strand=rep("+", 4),
gene_name=rep("TestGene1", 4));
names(test_exon_wide_gr) <- jamba::makeNames(rep("wide", length(test_exon_wide_gr)),
suffix="");
test_exon_wide_gr;
#> GRanges object with 4 ranges and 1 metadata column:
#> seqnames ranges strand | gene_name
#> <Rle> <IRanges> <Rle> | <character>
#> wide1 chr1 100-200 + | TestGene1
#> wide2 chr1 10300-10400 + | TestGene1
#> wide3 chr1 20500-20750 + | TestGene1
#> wide4 chr1 39900-40000 + | TestGene1
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengths
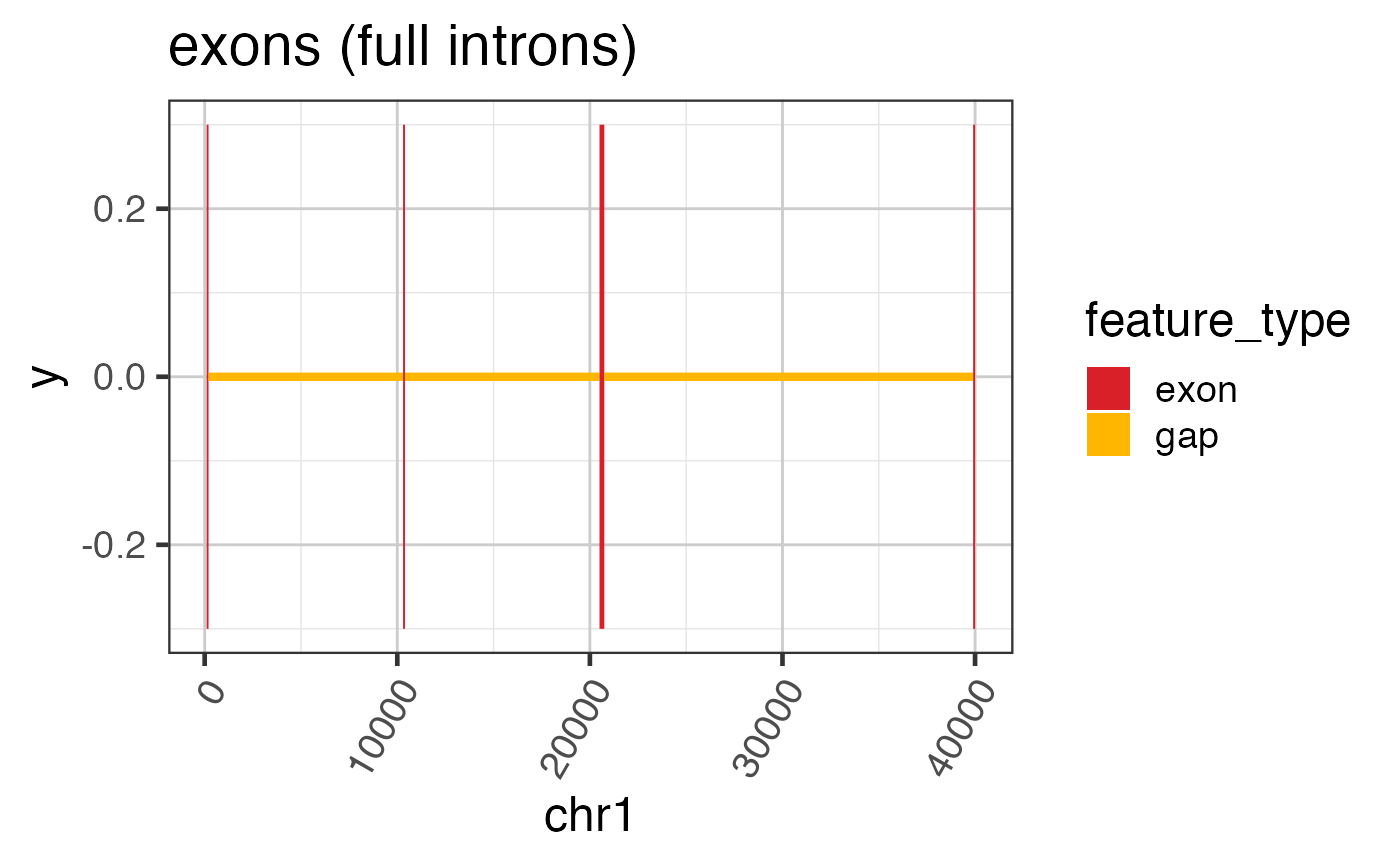
# To plot a simple GRanges object
widedf <- grl2df(test_exon_wide_gr);
ggWide <- ggplot(widedf, aes(x=x, y=y, group=id, fill=feature_type)) +
geom_polygon() +
colorjam::theme_jam() +
colorjam::scale_fill_jam() +
xlab("chr1") +
ggtitle("exons (full introns)")
print(ggWide);
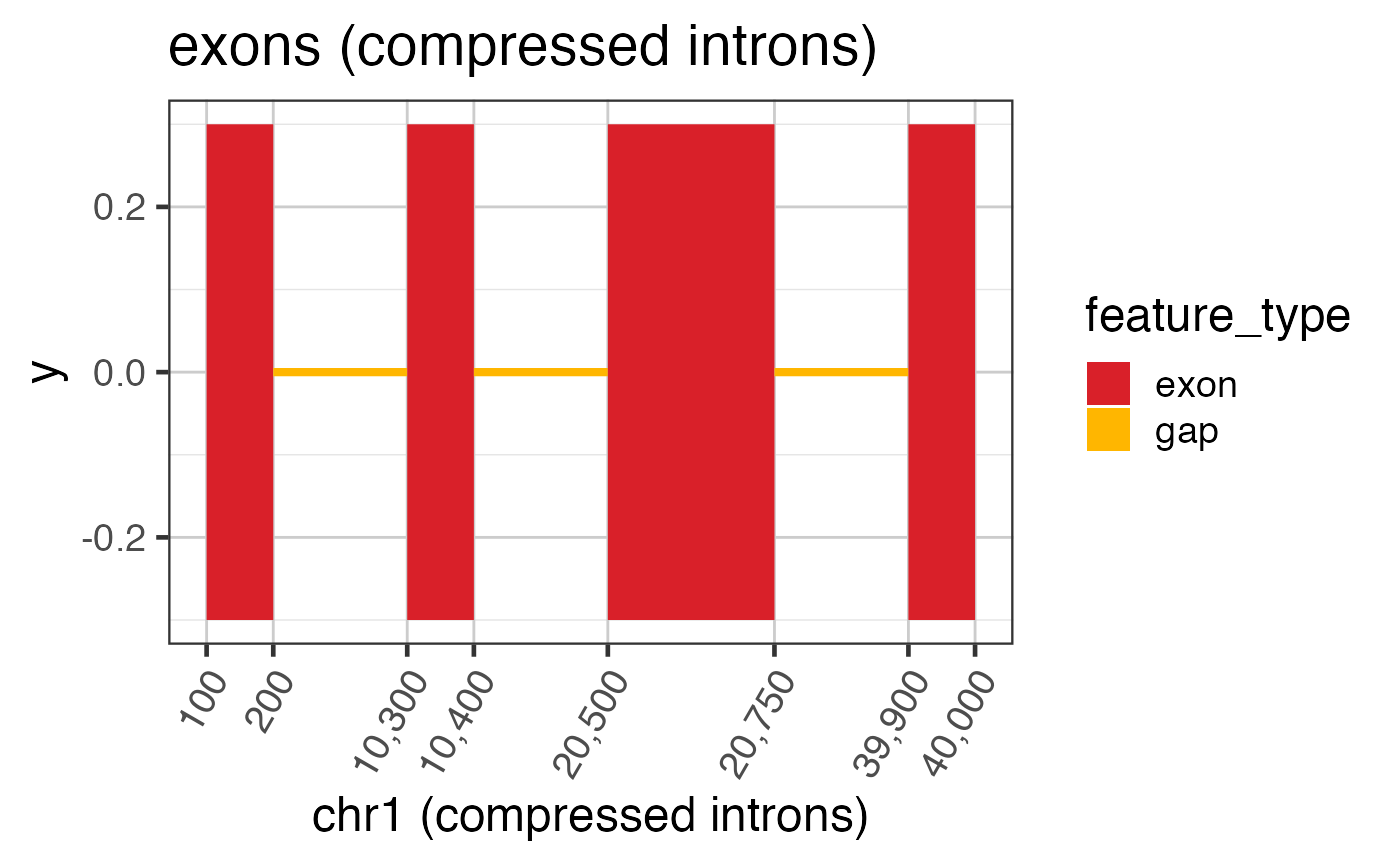
 # Now compress the introns keeping axis labels
ref2c <- make_ref2compressed(test_exon_wide_gr,
nBreaks=10);
ggWide2 <- ggWide +
scale_x_continuous(trans=ref2c$trans_grc) +
xlab("chr1 (compressed introns)") +
ggtitle("exons (compressed introns)")
print(ggWide2);
# Now compress the introns keeping axis labels
ref2c <- make_ref2compressed(test_exon_wide_gr,
nBreaks=10);
ggWide2 <- ggWide +
scale_x_continuous(trans=ref2c$trans_grc) +
xlab("chr1 (compressed introns)") +
ggtitle("exons (compressed introns)")
print(ggWide2);